Beim bis anhin gesunden Patienten kam es erstmals im Alter von 75 Jahren zu rezidivierenden entzündlichen Schüben. Die Symptome umfassten Fieber bis 40 °C, Nachtschweiss, Arthralgien bis Arthritiden (v.a. der MCP- und PIP-Gelenke), Hals-/Kieferschmerzen, Thrombophlebitiden und urtikarielle Hautausschläge mit jeweils ausgezeichnetem Ansprechen auf perorale Steroidstosstherapien (initiale Dosis: Prednison 70 mg täglich).
Im weiteren Verlauf kam es zusätzlich zu einer Thyreoiditis de Quervain sowie zu multiplen Lungenembolien. Laborchemisch fiel während den Schüben eine ausgeprägte humorale Entzündungsaktivität mit CRP-Erhöhung bis 209 mg/l und BSR >140 mm/h auf. Im Blutbild zeigten sich jeweils eine makrozytäre Anämie sowie Leukopenie.
Trotz umfangreicher Diagnostik inkl. Autoimmunserologien, infektiologischer Abklärung sowie Bildgebungen inkl. PETCT gelang es nicht, die Symptomatik einer spezifischen Krankheitsentität zuzuordnen. Es wurde daher von einem undifferenzierten systemischen autoinflammatorischen Syndrom ausgegangen. Aufgrund des kontinuierlichen Glukokortikoid-Bedarfs war es mittlerweile leider zur Manifestation eines Diabetes Typ 2 gekommen. Es wurde versucht, eine steroidsparende Basistherapie mit Methotrexat s.c. zu etablieren; bei fehlendem Ansprechen wurde zusätzlich Tocilizumab i.v. begonnen.
Dennoch kam es unter Reduktion der Steroidtherapie zu rezidivierenden Krankheitsschüben. Zudem ergaben sich zwischenzeitlich neue Hautbefunde so konnte in einer Hautbiopsie eine leukozytoklastische
Vaskulitis gesichert werden; in einer bronchoalveolären Lavage waren zudem alveoläre Hämorrhagien nachweisbar. MR-graphisch zeigten sich myositische Veränderungen der Beckenmuskulatur mit Ödem und Kontrastmittelanreicherung. Im CT-Thorax waren Lungenembolien zu sehen (➞ siehe Abbildung 1).
Aufgrund der bis anhin ungeklärten Genese und des therapierefraktären Verlaufs wurde eine paraneoplastische Genese in Betracht gezogen. Tatsächlich kam es 3 Jahre nach Erstmanifestation erstmals zum Nachweis einer klonalen B-Zell-Expansion im peripheren Blut. Eine anschliessende Knochenmarkspunktion stellte die Diagnose eines kleinzelligen lymphozytischen Lymphoms (SLL). Es wurde eine Chemotherapie mit Rituximab und Cyclosphosphamid initiiert.
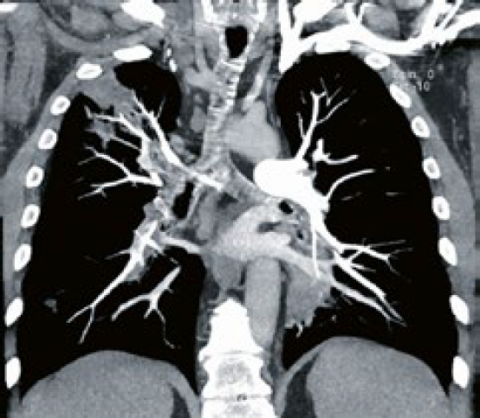
Abbildung 1: CT-Thorax: Zentrale Lungenembolien rechts und periphere Lungenembolien links mit Infarktpneumonien
Im Anschluss an die Chemotherapie und weiterhin verabreichter Prednison-Therapie p.o. war die entzündliche Symptomatik zunächst gut kontrolliert. Sobald jedoch Prednison auf <15 mg täglich reduziert wurde, kam es erneut zu entzündlichen Symptomen. Da somit das autoinflammatorische Syndrom auch unter kausaler Behandlung des Malignoms nicht rückgängig war, wurde nun eine neue Therapie mit Colchicin etabliert. Leider konnte hiermit ebenfalls keine anhaltende Krankheitskontrolle erreicht werden. Infolgedessen wurde eine Therapie mit Rituximab begonnen, worunter sich allerdings abermals kein Ansprechen abzeichnete.
Im Rahmen einer erneuten Standortbestimmung konnte bronchoskopisch eine kryptogen organisierende Pneumonie (COP) diagnostiziert werden. Aufgrund einer T-Zell-Prädominanz in der Zytologie der BAL wurde infolge pathophysiologischer Überlegungen eine Therapie mit Abatacept etabliert. Leider kam es auch hierunter zu keinerlei Ansprechen. Gleichzeitig verhielt sich die Erkrankung weiterhin äusserst steroidsensibel und es waren kontinuierliche Prednison-Dosen von mind. 15 mg/d notwendig. Es erfolgte ein Therapiewechsel auf Cyclophosphamid, worunter nach 3 Zyklen weiterhin keinerlei Ansprechen absehbar war und wiederholt Prednison-Dosen bis 80 mg/d aufgrund schwerer entzündlicher Schübe mit Hospitalisationsbedarf notwendig waren. Zuletzt waren jeweils tägliche Prednison-Dosen von 20–25 mg p.o. notwendig, da es bei Unterschreiten dieser Schwelle zu erneuten inflammatorischen Schüben kam.
Aufgrund des hartnäckigen und ungewöhnlichen Verlaufs erfolgte ein erneutes diagnostisches Work-Up mit Studium der aktuellen Literatur. Hierbei kam der Verdacht eines VEXAS-Syndroms auf. Es erfolgte daher eine erneute Analyse des früheren Knochenmarksaspirats, welches bei Erstmanifestation untersucht wurde. Tatsächlich zeigten sich hierbei vakuolisierte myeloische und erythrozytäre Vorstufen. Eine zusätzliche genetische Analyse aus dem Knochenmark stellte den Nachweis einer Mutation im UBA1 Gen (c. 121A>G, p.Met41Val), so dass die Diagnose eines VEXAS-Syndroms gestellt werden konnte.
Diskussion
Bei diesem Patienten mit episodisch auftretenden inflammatorischen Schüben konnte nach mehrjähriger Krankheitsgeschichte mit zunehmenden Krankheitsmanifestationen, Notwendigkeit einer Dauer Glukokortikoidtherapie und multiplen frustranen Versuchen einer Basistherapie die Diagnose eines VEXAS-Syndroms gestellt werden. Interessanterweise war das Krankheitsbild bei Symptombeginn unseres Patienten noch nicht bekannt, da das Syndrom erst 2020 in der Literatur beschrieben wurde. Retrospektiv präsentierte sich unser Patient äusserst typisch für die entsprechende Krankheitsentität: hierzu passten nicht nur die schubartigen inflammatorischen Symptome mit deutlich erhöhter Akut-Phase-Reaktion, sondern auch die hämatologischen Befunde inklusive Nachweis einer lymphoproliferativen Erkrankung, die rezidivierenden thrombembolischen Ereignisse, pulmonale sowie kutane Beteiligung. Typisch gestaltete sich auch die ausgeprägte Glukokortikoid-Sensibilität mit fehlendem Ansprechen auf multiple steroidsparende Basistherapien.
Das VEXAS-Syndrom
Beim VEXAS-Syndrom handelt es sich um ein autoinflammatorisches Syndrom, welches erstmals im Dezember 2020 durch Beck et al. Im NEJM beschrieben wurde.1 Die Autor*innen beschrieben ein Syndrom, welches sie bei 25 Männern in höherem Lebensalter (medianes Alter bei Krankheitsbeginn: 64 J., (45–80 J.)) und mit autoinflammatorischem Krankheitsbild identifizieren konnten. Alle Patienten wiesen ein fehlendes Ansprechen auf multiple konventionelle und biologische antirheumatische Therapien auf, u.a. Methotrexat, Anakinra, Mycophenolat mofetil, Colchicin, Tocilizumab und TNF-a Blocker. Zur Kontrolle der inflammatorischen Aktivität zeigten sich lediglich Glukokortikoide wirksam, häufig in höheren Dosen. Hierbei gelang der molekulargenetische Nachweis einer somatischen Mutation des UBA1-Gens. Somatische Mutationen betreffen im Gegensatz zu Keimbahnmutationen nicht die Keimzellen, sondern somatische Zellen (Körperzellen). Entsprechend werden somatische Mutationen nicht an die nachfolgende Generation vererbt. Das UBA1-Gen kodiert für das Enzym Ubiquitin-aktivierendes Enzym 1, UBA1, auch E1 genannt), welches eine wesentliche Rolle in der Ubiquitinierung des Proteasoms einnimmt.2 Da das UBA1-Gen auf dem X-Chromosom lokalisiert ist, sind häufiger Männer von der Erkrankung betroffen, da Männer in der Regel nur eine Kopie von X-chromosomalen Genen aufweisen.1,6
| Organsystem | Manifestation |
|---|---|
| Konstitutionell | Fieber, Lymphadenopathie, Fatigue, Gewichtsverlust |
| Hämatologisch | Makrozytäre Anämie, Thrombozytopenie, Myelodysplastisches Syndrom |
| Haut | Neutrophile Dermatose, kutane Vaskulitis |
| Bewegungsapparat | Arthralgien, Arthritis, (Myositis) |
| Knorpel | Chondritis (aurikulär, nasal) |
| Gefässe | Vaskulitis (kleine, mittlere oder grosse Gefässe), Thrombembolien |
| Lunge | Alveolitis, Pleuritis, NSIP, COP, Bronchiolitis obliterans |
| Weitere (selten) | gastrointestinal, kardial, neurologisch |
Tabelle 1: Klinische Manifestationen bei VEXAS-Syndrom
(UBA1, auch E1 genannt), welches eine wesentliche Rolle in der Ubiquitinierung des Proteasoms einnimmt.2 Da das UBA1-Gen auf dem X-Chromosom lokalisiert ist, sind häufiger Männer von der Erkrankung betroffen, da Männer in der Regel nur eine Kopie von X-chromosomalen Genen aufweisen.1,6 Im Knochenmark von Erkrankten können Vakuolen in myeloischen und erythrozytären Vorläuferzellen nachgewiesen werden, elektronenmikroskopisch bestehend aus Lipidtröpfchen und Zellorganellen, einschliesslich degenerierter Mitochondrien. Die Erkrankung wurde von den Autor*innen VEXAS Syndrom genannt. Das Akronym «VEXAS» steht für: vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic.
Klinik des VEXAS-Syndroms
Das VEXAS-Syndrom führt zu einer Vielzahl von entzündlichen Symptomen mit Multiorganbeteiligung. Typisch sind rezidivierendes Fieber mit laborchemisch erhöhter Akut-Phase-Reaktion (CRP, BSR), hämatologische Auffälligkeiten (makrozytäre Anämie, Thrombozytopenie), thrombembolische Ereignisse, Chondritis, eine kutane sowie pulmonale Beteiligung.1 Eine Myositis, wie sie bei vorliegendem Patienten nachweisbar war, ist nicht typisch, wird aber in der Literatur vereinzelt beschrieben.2^12,13 Gehäuft liegt auch eine hämatologische Neoplasie vor. Eine Untersuchung aus dem Jahre 2021 von Obiorah et al. an 16 Patient*innen zeigte in 62 % den Nachweis einer hämatologischen Begleiterkrankung, u.a. Myelodysplastisches Syndrom (MDS), Multiples Myelom, MGUS, monoklonale B-Zell-Lymphozytosen.1,3 Zu erwähnen ist, dass die klinischen Manifestationen nicht spezifisch für die Erkrankung sind und mit einer Vielzahl von anderen rheumatisch-entzündlichen Erkrankungen interferieren. So erfüllen die Patient*innen teilweise Diagnose- oder Klassifikationskriterien für andere Krankheitsentitäten wie Relapsing Polychondritis, Sweet-Syndrom, Polyarteriitis nodosa oder Riesenzellarteriitis. Eine Übersicht der relevanten klinischen Manifestationen findet sich folgend in Tabelle 1.1,5,6,11
Diagnostik beim VEXAS-Syndrom
An ein VEXAS-Syndrom solltevor allem gedacht werden bei Vorliegen folgender Charakteristika:4,5
- Männliche Patienten ab der 5. Lebensdekade
- Klinisches Bild einer autoinflammatorischen Erkrankung (➞ siehe Tabelle 1)
- Hämatologische Auffälligkeiten (makrozytäre Anämie, Thrombozytopenie, MDS)
- Therapierefraktärer Verlauf
Im rheumatologischen Alltag sollte insbesondere auch bei Vorliegen einer rheumatisch-entzündlichen Erkrankung mit therapierefraktärem Verlauf ein VEXAS-Syndrom in Betracht gezogen werden. So kann das Bild einer Relapsing Polychondritis als Manifestation eines VEXAS-Syndroms auftreten.8,9,10 Doch auch bei Phänotyp einer Polyarteriitisnodosa, Riesenzellarteriitis oder eines Sweet-Syndroms sollte bei hartnäckigem Verlauf differentialdiagnostisch ein VEXAS-Syndrom erwogen werden.1,5,8 Besteht die klinische Verdachtsdiagnose, kann eine Knochenmarksbiopsie das Vorliegen von zytoplasmatischen Vakuolen in myeloischen und erythrozytären Vorläuferzellen nachweisen (➞ siehe Abbildung 2). Die definitive Diagnose bedarf einer molekulargenetischen Analyse mit Nachweis einer pathognomischen Mutation im UBA1-Gen.7,11 (➞ siehe Abbildung 2).
| V | Vakuolen |
| E | E1-Enzym |
| X | X-chromosomal |
| A | Autoinflammatorisch |
| S | Somatische Mutation |
Therapie und Verlauf des VEXAS-Syndroms Aufgrund des erst neu beschriebenen Krankheitsbildes, lediglich kleiner Patient*innen-Populationen sowie mangels randomisiert-kontrollierter Studien existieren (noch) keine etablierten Therapieprotokolle oder Leitlinien. Aktuelle Empfehlungen beruhen auf Erfahrungswerten, pathophysiologischen Überlegungen sowie Case Reports.5,14,15 Glukokortikoide führen bekannterweise zu einem guten Ansprechen, wobei häufig leider höhere Steroiddosen (Prednison
20–40 mg) notwendig sind, um die inflammatorische Aktivität in Schach zu halten.1,11 Als steroidsparende Basistherapien finden sich positive Daten zu Tocilizumab, dem JAK-Inhibitor Ruxolitinib sowie zu IL-1-Antagonisten (Anakinra/Canakimumab). Zu beachten ist, dass unter Anakinra ungewöhnlich schwere lokale Hautreaktionen beschrieben wurden, die zu Therapieabbrüchen führen mussten.16,17 Innerhalb der onkologischen Therapien finden sich günstige
Daten für den Antimetaboliten Azacitidin – insbesondere bei hämatologischer Beteiligung/assoziiertem MDS.
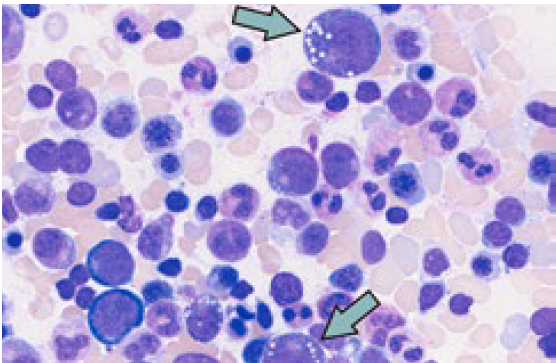
Abbildung 2: Knochenmarkspunktion: Nachweis von Vakuolen in myeolischen Zellen. (Quelle: Hämatologische Diagnostik, Nadija Wegener USZ)
Zusätzlich finden sich Case Reports mit positivem Ansprechen
auf eine allogene Stammzelltransplantation,
wobei hier jedoch sicherlich das Nutzen-Risiko-Verhältnis
abgewogen werden muss.5,11,15 Zu IVIG findet sich ein einzelner
Fallbeschrieb bei einem Patienten mit zusätzlicher
Spondyloarthritis und unter anti-IL-17 Therapie.18
Therapie bei unserem Patienten
Bei unserem Patienten wurde nach Diagnosestellung eine Therapie mit dem JAK-Inhibitor Ruxolitinib p.o. etabliert, welches aktuell aufdosiert wird und worunter die Glukokortikoid-Therapie aktuell auf Prednison 12.5 mg/d reduziert werden konnte, während zuvor Dosen von 20– 25 mg/d notwendig waren.
Take Home Messages
- Beim VEXAS-Syndrom handelt es sich um ein autoinflammatorisches Syndrom auf Basis einer Mutation im X-chromosomalen UBA1-Gen.
- In der Knochenmarksbiopsie können zytoplasmatische Vakuolen in myeloischen und erythrozytären Vorläuferzellen nachweisen werden
- Bei hartnäckigem Verlauf rheumatisch-entzündlicher Manifestationen trotz multiplen Therapien sollte v.a. bei Männern ab der 5. Lebensdekade an ein VEXAS-Syndrom gedacht werden.
- Verhält sich der Verlauf einer Erkrankung hartnäckig oder atypisch, lohnt sich eine erneute Standortbestimmung unter Berücksichtigung der aktuellen Literatur.
Abkürzungen
BAL Bronchoalveoläre Lavage
COP Kryptogen organisierende Pneumonie
IVIG Intravenöse Immunglobuline
JAK Januskinase
MDS Myelodysplastisches Syndrom
MGUS Monoklonale Gammopathie unklarer Signifikanz
NEJM The New England Journal of Medicine
NSIP Nonspecific interstitial pneumonia
UBA1 Ubiquitin-aktivierendes Enzym 1
- Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC,
Pei W, Balanda N, Ross DL, Ospina Cardona D, Wu Z, Patel
B, Manthiram K, Groarke EM, Gutierrez-Rodrigues F, Hoffmann
P, Rosenzweig S, Nakabo S, Dillon LW, Hourigan CS,
Tsai WL, Gupta S, Carmona-Rivera C, Asmar AJ, Xu L, Oda
H, Goodspeed W, Barron KS, Nehrebecky M, Jones A, Laird
RS, Deuitch N, Rowczenio D, Rominger E, Wells KV, Lee CR,
Wang W, Trick M, Mullikin J, Wigerblad G, Brooks S, Dell›Orso
S, Deng Z, Chae JJ, Dulau-Florea A, Malicdan MCV, Novacic
D, Colbert RA, Kaplan MJ, Gadina M, Savic S, Lachmann HJ,
Abu-Asab M, Solomon BD, Retterer K, Gahl WA, Burgess
SM, Aksentijevich I, Young NS, Calvo KR, Werner A, Kastner
DL, Grayson PC. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. 2020
Dec 31;383(27):2628-2638. doi: 10.1056/NEJMoa2026834. Epub
2020 Oct 27. PMID: 33108101; PMCID: PMC7847551. - Molekulare Genetik, Rolf Knippers, 9. Auflage, Thieme, ISBN 10:
3-13-477009-1, ISBN 13: 978-3-13-477009-4 - Obiorah IE, Patel BA, Groarke EM, Wang W, Trick M, Ombrello
AK, Ferrada MA, Wu Z, Gutierrez-Rodrigues F, Lotter J, Wilson
L, Hoffmann P, Cardona DO, Patel N, Dulau-Florea A, Kastner
DL, Grayson PC, Beck DB, Young NS, Calvo KR. Benign
and malignant hematologic manifestations in patients with
VEXAS syndrome due to somatic mutations in UBA1. Blood
Adv. 2021 Aug 24;5(16):3203-3215. doi: 10.1182/bloodadvances.
2021004976. PMID: 34427584; PMCID: PMC8405186. - Grayson PC, Patel BA, Young NS. VEXAS syndrome. Blood.
2021 Jul 1;137(26):3591-3594. doi: 10.1182/blood.2021011455.
PMID: 33971000; PMCID: PMC8462403. - Kobak S. VEXAS syndrome: Current clinical, diagnostic
and treatment approaches. Intractable Rare Dis Res. 2023
Aug;12(3):170-179. doi: 10.5582/irdr.2023.01020. PMID:
37662628; PMCID: PMC10468411. - Zhang Y, Dong X, Wang H. VEXAS Syndrome-Review. Glob Med
Genet. 2023 Jul 10;10(3):133-143. doi: 10.1055/s-0043-1770958.
PMID: 37501758; PMCID: PMC10370470. - Vitale A, Caggiano V, Bimonte A, Caroni F, Tosi GM, Fabbiani A,
Renieri A, Bocchia M, Frediani B, Fabiani C, Cantarini L. VEXAS
syndrome: a new paradigm for adult-onset monogenic autoinflammatory
diseases. Intern Emerg Med. 2023 Apr;18(3):711-
722. doi: 10.1007/s11739-023-03193-z. Epub 2023 Jan 20. PMID:
36662445; PMCID: PMC10082120. - Bourbon E, Heiblig M, Gerfaud Valentin M, Barba T, Durel CA,
Lega JC, Barraco F, Sève P, Jamilloux Y, Sujobert P. Therapeutic
options in VEXAS syndrome: insights from a retrospective
series. Blood. 2021 Jul 1;137(26):3682-3684. doi: 10.1182/
blood.2020010177. PMID: 33619558. - Khitri MY, Guedon AF, Georgin-Lavialle S, Terrier B, Saadoun D,
Seguier J, le Besnerais M, De Moreuil C, Denis G, Gerfaud-Valentin
M, Allain JS, Maria A, Bouillet L, Grobost V, Galland J,
Kosmider O, Dumont A, Devaux M, Subran B, Schmidt J, Marianetti-
Guingel P, Audia S, Palat S, - McHugh J. Pathogenic UBA1 variants define a subset of relapsing
polychondritis. Nat Rev Rheumatol. 2021 Jun;17(6):312. doi:
10.1038/s41584-021-00624-z. PMID: 33941913.Roux-Sauvat M,
Jachiet V, Hirsch P, Fain O, Mekinian A; French VEXAS group
and MINHEMON. Comparison between idiopathic and VEXAS-
relapsing polychondritis: analysis of a French case series of
95 patients. RMD Open. 2022 Jul;8(2):e002255. doi: 10.1136/rmdopen-
2022-002255. PMID: 35868738; PMCID: PMC9315905. - Koster MJ, Samec MJ, Warrington KJ. VEXAS Syndrome-A
Review of Pathophysiology, Presentation, and Prognosis.
J Clin Rheumatol. 2023 Sep 1;29(6):298-306. doi: 10.1097/
RHU.0000000000001905. Epub 2022 Oct 17. PMID: 36251488. - Topilow JS, Ospina Cardona D, Beck DB, Ferrada MA, Mc-
Mahan ZH, Paik JJ. Novel genetic mutation in myositis-variant
of VEXAS syndrome. Rheumatology (Oxford). 2022 Nov
28;61(12):e371-e373. doi: 10.1093/rheumatology/keac356. PMID:
35713495; PMCID: PMC9707058. - Matsuki Y, Kawai R, Suyama T, Katagiri K, Kanazawa N, Inaba
Y. A case of VEXAS syndrome with myositis possibly associated
with macrophage activation syndrome. J Dermatol. 2022
Dec;49(12):e441-e443. doi: 10.1111/1346-8138.16535. Epub 2022
Aug 4. PMID: 35924490. - Heiblig M, Patel BA, Groarke EM, Bourbon E, Sujobert P. Toward
a pathophysiology inspired treatment of VEXAS syndrome.
Semin Hematol. 2021 Oct;58(4):239-246. doi: 10.1053/j.seminhematol.
2021.09.001. Epub 2021 Oct 5. PMID: 34802546. - Boyadzhieva Z, Ruffer N, Kötter I, Krusche M. How to treat VEXAS-
Syndrome: a systematic review on effectiveness and safety
of current treatment strategies. Rheumatology (Oxford). 2023
May 26:kead240. doi: 10.1093/rheumatology/kead240. Epub
ahead of print. PMID: 37233149. - van der Made CI, Potjewijd J, Hoogstins A, Willems HPJ, Kwakernaak
AJ, de Sevaux RGL, van Daele PLA, Simons A, Heijstek
M, Beck DB, Netea MG, van Paassen P, Elizabeth Hak A, van
der Veken LT, van Gijn ME, Hoischen A, van de Veerdonk FL,
Leavis HL, Rutgers A. Adult-onset autoinflammation caused by
somatic mutations in UBA1: A Dutch case series of patients with
VEXAS. J Allergy Clin Immunol. 2022 Jan;149(1):432-439.e4. doi:
10.1016/j.jaci.2021.05.014. Epub 2021 May 25. PMID: 34048852. - Staels F, Betrains A, Woei-A-Jin FJSH, Boeckx N, Beckers
M, Bervoets A, Willemsen M, Neerinckx B, Humblet-Baron S,
Blockmans DE, Vanderschueren S, Schrijvers R. Case Report:
VEXAS Syndrome: From Mild Symptoms to Life-Threatening
Macrophage Activation Syndrome. Front Immunol. 2021 Apr
23;12:678927. doi: 10.3389/fimmu.2021.678927. Erratum in:
Front Immunol. 2021 Aug 10;12:748756. PMID: 34046042; PMCID:
PMC8147557. - Magnol M, Couvaras L, Degboé Y, Delabesse E, Bulai-Livideanu
C, Ruyssen-Witrand A, Constantin A. VEXAS syndrome in a patient
with previous spondyloarthritis with a favourable response
to intravenous immunoglobulin and anti-IL17 therapy. Rheumatology
(Oxford). 2021 Sep 1;60(9):e314-e315. doi: