Die schwerwiegendste klinische Manifestation der hypertrophen Kardiomyopathie ist der plötzliche Herztod, welcher bei diesen Patienten und Patientinnen durchschnittlich doppelt so häufig auftritt wie in der Normalbevölkerung. Die Hälfte der Patienten und Patientinnen mit einer HCM haben keine oder nur sehr milde Symptome. Die hypertrophe Kardiomyopathie ist zwar nicht heilbar, aber gut zu behandeln – mit Medikamenten, interventionellen oder chirurgischen Behandlungen.
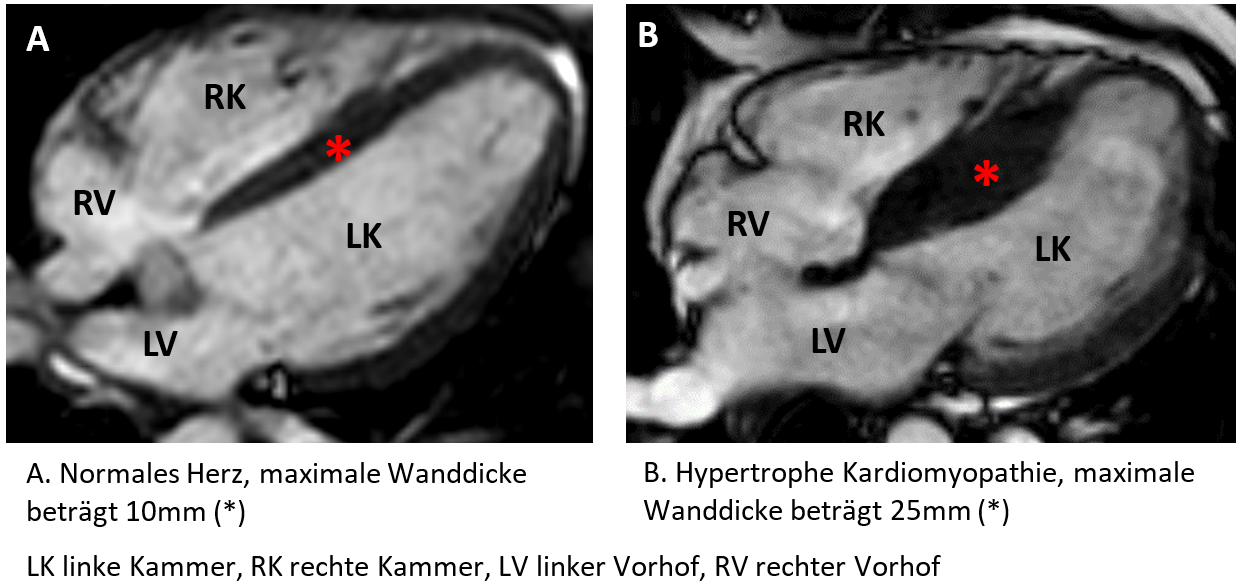
Die hypertrophe Kardiomyopathie ist eine meist genetisch bedingte Herzerkrankung, bei der sich die Muskelzellen in der Herzwand vergrössern und der Herzmuskel (Myokard) verdickt. In den meisten Fällen ist die linke Herzkammer betroffen, welche bis zu zwei bis vier Mal dicker sein kann im Vergleich zu einem normalen Herzmuskel.
Hypertrophe Kardiomyopathie – Häufigkeit und Alter
Im Gegensatz zu vielen anderen Herz-Krankheiten ist die hypertrophe Kardiomyopathie eher selten: Die Häufigkeit liegt bei 0.2% in der Gesamtbevölkerung und kann sich in jedem Lebensalter manifestieren.
Hypertrophe Kardiomyopathie - Ursachen liegen oft in den Genen
Die Ursachen der hypertrophen Kardiomyopathie sind in vielen Fällen fehlerhafte Gene. Wir schätzen, dass rund 40 bis 60 Prozent der Heranwachsenden und Erwachsenen mit HCM eine Erbgutveränderung im kardialen Sarkomerprotein-Gen aufweisen. Dieses Gen liefert die Bauanleitung für die Sarkomere, die kleinsten Einheiten der Muskeln. Ist es verändert, weichen anschliessend die Muskelzellen und auch die Struktur des Herzmuskels vom Normalfall ab. Die Muskelzellen vergrössern sich und die Herzwände werden dicker.
Der Erbgang ist autosomal-dominant. Das bedeutet, dass die Veränderung nur auf einem der beiden Gene der Eltern vorhanden sein muss, damit ein Kind erkrankt (es bekommt jedes Chromosom zweimal – einmal vom Vater, einmal von der Mutter). Die Wahrscheinlichkeit, dass ein Kind das „kranke“ Gen erbt, liegt demnach bei 50 Prozent.
Auch wenn die hypertrophe Kardiomyopathie meist genetisch bedingt ist und familiär gehäuft vorkommt, können die verschiedenen Familienmitglieder die Erkrankung aber in ganz unterschiedlicher Ausprägung ausbilden. Die Bandbreite reicht von fast normal bis zu einer ausgeprägten Verdickung.
Hypertrophe Kardiomyopathie – andere Ursachen
Daneben gibt es noch seltenere Gendefekte sowie nicht-genetische Ursachen, die zu einer Verdickung des Herzmuskels führen und sich ähnlich wie eine hypertrophe Kardiomyopathie präsentieren kann. Bei rund 25 bis 30 Prozent der Patienten und Patientinnen mit hypertropher Kardiomyopathie sind unbekannte Gene verändert (mutiert). Bei etwa fünf bis zehn Prozent der Erwachsenen sind noch andere Krankheiten und Faktoren beteiligt:
- Angeborene Erkrankungen des Stoffwechsels, z.B. Glykogenspeicherkrankheiten, Carnitin-Erkrankungen, lysosomale Speicherkrankheiten oder Morbus Anderson Fabry
- Angeborene Erkrankungen der Muskeln und Nerven (neuromuskuläre Erkrankungen), z.B. die Friedreich-Ataxie
- Mitochondriale Erkrankungen, z.B. das MELAS-Syndrom
- Malformationssyndrome, z.B. Noonan- oder LEOPARD-Syndrom
- Amyloidose
- Fettleibigkeit
- Medikamente
Da die korrekte Diagnose Auswirkungen auf die Prognose und Therapie haben kann, legen wir viel Wert auf den diagnostischen Prozess.
Hypertrophe Kardiomyopathie – Symptome und zu erwartende Komplikationen
Die Symptome bei einer hypertrophen Kardiomyopathie hängen davon ab, wie schwer die Erkrankung ausgeprägt ist. Manche verspüren nur milde Symptome, während die hypertrophe Kardiomyopathie bei anderen ausgeprägtere Beschwerden verursacht. Diese können den Alltag, die Lebensfreude und die Lebensqualität massgeblich beeinträchtigen.
- Abnahme der körperlichen Leistungsfähigkeit, Schwäche
- Atemnot bei körperlicher Anstrengung
- Engegefühl, Druckgefühl oder Schmerzen in der Brust bei Anstrengung oder in Ruhe
- Unregelmässiger Puls (Galopp), Herzklopfen, Herzrasen (Palpitationen)
- Schwindel
- Ohnmachtsanfälle, Bewusstlosigkeit
- Im schlimmsten Fall: plötzlicher Herztod
Patienten und Patientinnen mit hypertropher Kardiomyopathie können im Krankheitsverlauf verschiedene Komplikationen entwickeln.
- Einengung des Ausflusstrakts der linken Kammer (obstruktive Form), die zu einer Verminderung des Blutvolumens führt, welches dem Körper für seine Funktionen zur Verfügung gestellt wird. Klinisch zeigt sich dies meist in einer Abnahme der körperlichen Leistungsfähigkeit. Ungefähr 70% der Patientinnen und Patienten können eine solche Einengung des Ausflusstrakts entwickeln.
- Steife linke Herzkammer (‚diastolische Dysfunktion‘): die Verdickung des Herzmuskels führt dazu, dass die linke Kammer eher steif ist und sich in der Füllungsphase, wenn das Herz das Blut aus dem Körperkreislauf aufnimmt, nicht so gut entspannen kann. Dadurch steigen die Drücke im Herz an, was einerseits mit Atemnot und andererseits mit einem Druckgefühl in der Brust einhergehen kann.
- Herzschwäche: wenige Patienten und Patientinnen (5-10%) können im Verlauf der Krankheit eine Abnahme der Pumpfunktion entwickeln, die dann zu einer Herzschwäche führt. Dies kann mit Wassereinlagerungen in den Beinen und in der Lunge (Atemnot, Husten im Liegen) einhergehen oder ganz allgemein mit Müdigkeit und Abnahme der Leistungsfähigkeit.
- Vorhofflimmern: hierbei handelt es sich um eine Rhythmusstörung die aus dem linken Vorhof kommt und meist mit einem raschen, unregelmässigen Puls einhergeht. Diese Rhythmusstörung ist nicht lebensbedrohlich, führt aber meist zu deutlichen Symptomen. Von grösster Wichtigkeit ist der Beginn einer Blutverdünnung, damit sich im Herzen keine Gerinnsel bilden können, die im schlimmsten Fall einen Schlaganfall verursachen könnten. Zirka ein Drittel der Patienten und Patientinnen mit hypertropher Kardiomyopathie entwickeln im Verlauf der Krankheit Vorhofflimmern, was zu Beginn meist nur kurz andauert und im Extremfall aber permanent bleiben kann.
- Plötzlicher Herztod: dabei handelt es sich um die schwerwiegendste Komplikation, die aber glücklicherweise selten auftritt. Die Ereignisrate bei Patienten und Patientinnen mit hypertropher Kardiomyopathie liegt bei 0.6% pro Jahr, im Vergleich zu 0.3% pro Jahr bei der Normalbevölkerung. Diese Rhythmusstörung kündigt sich nicht im Voraus an und kann aus dem absoluten Wohlbefinden heraus auftreten und führt zum Kreislaufkollaps der zum Tod führt, wenn nicht rasch Erste Hilfe vor Ort ist
Hypertrophe Kardiomyopathie: Behandlungsmöglichkeiten und Verhaltensmassnahmen
An der Behandlung der hypertrophen Kardiomyopathie sind Ärzte und Ärztinnen unterschiedlicher Fachdisziplinen beteiligt. Zentral ist der klinische Kardiologe oder die klinische Kardiologin, welche die Patienten und Patientinnen in der Sprechstunde sieht und dann je nachdem, welches Hauptproblem vorliegt mit den entsprechenden Spezialisten und Spezialistinnen aus den anderen Fachgebieten die interdisziplinäre Behandlung und Betreuung sucht. Die hypertrophe Kardiomyopathie ist nicht heilbar. Wir können die Beschwerden jedoch gut behandeln und das Risiko für Komplikationen wie den plötzlichen Herztod senken.
Folgende Behandlungsmöglichkeiten gibt es:
- Einengung des Ausflusstrakts der linken Kammer: hier kommen in erster Linie Medikamente zum Einsatz, die das Herz entlasten sollen und den Blutfluss aus dem Herzen in den Kreislauf erleichtern sollen. Beispiele sind Betablocker (Bisoprolol, Metoprolol, etc.) oder Kalziumantagonisten (Verapamil, Diltiazem). Wenn die Medikamente nicht ausreichen, kommen invasivere Methoden zum Einsatz, mit dem Ziel, im Ausflusstrakt der linken Kammer das verdickte Muskelgewebe zu verdünnen, damit das Blut wieder ungehindert fliessen kann. Hierzu gehört die Septalalkoholablation, bei welcher der Herzkatheter-Spezialist oder die Herzkatheter-Spezialistin Alkohol in die kleinen arteriellen Blutgefässe spritzt, welche die Herzscheidewand versorgen und dadurch das verdickte Gewebe verödet. Eine weitere Möglichkeit stellt die chirurgische Myektomie dar, bei welcher der Herzchirurg oder die Herzchirurgin einen Teil des verdickten Herzmuskels aus dem Ausflusstrakt mit dem Skalpell entfernt.
- Steife linke Herzkammer (‚diastolische Dysfunktion‘): Hier kommen primär Medikamente zur Anwendung und zwar auch Betablocker oder Kalziumantagonisten.
- Herzschwäche: Hier kommen Medikamente zur Anwendung, die auch bei anderen Formen der Herzschwäche angewendet werden, mit dem Ziel, das Herz zu entlasten und die Pumpfunktion solange wie möglich zu stabilisieren. Im Extremfall kann die Behandlung bei diesen Formen bis zur Herztransplantation gehen, was aber bei Patienten und Patientinnen mit hypertropher Kardiomyopathie sehr, sehr selten der Fall ist.
- Vorhofflimmern: hier versuchen wir, entweder durch Medikamente oder elektrisch durch Kardioversion (Applikation von Strom im Kurzschlaf) das Herz wieder in den normalen Rhythmus zu bringen. Im Anschluss werden meist Rhythmusstabilisierende Medikamente als Dauertherapie verabreicht. Von grösster Wichtigkeit ist der Beginn einer dauerhaften Blutverdünnung zur Vermeidung der Bildung von Blutgerinnseln im Herzen und Schlaganfallprophylaxe. Wenn sich die Vorhofflimmernepisoden häufen, kann auch eine Katheter-gesteuerte Verödung der Rhythmusstörung diskutiert werden.
- Plötzlicher Herztod: Das Risiko eines plötzlichen Herztodes wird für jeden Patienten und für jede Patientin bei jedem Sprechstundentermin neu beurteilt. Wird das Risiko als hoch eingeschätzt, empfehlen wir die Implantation eines implantierbaren Defibrillators (ICD von engl. implantable cardioverter defibrillator). Dieser kann das Auftreten der Rhythmusstörung nicht verhindern und vermindert auch nicht die Symptome. Aber er fungiert als Schutzengel und gibt bei Auftreten der potentiell tödlichen Rhythmusstörung von innen einen elektrischen Schock ab, um die Rhythmusstörung zu beenden. Dies nehmen die Patienten und Patientinnen in der Regel nicht wahr, weil sie in diesem Moment häufig das Bewusstsein verloren haben.
- Sportempfehlungen: wir empfehlen moderaten Ausdauersport mit Aufwärm- und Auslaufphase. Ideal sind Laufen, Joggen, Velofahren, Schwimmen, etc. Was Patienten und Patientinnen mit hypertropher Kardiomyopathie schlecht tolerieren sind Sportarten, die mit Sprints verbunden sind (z.B. Squash, Badminton, Einzeltennis). Ebenfalls raten wir von intensivem Krafttraining ab; es sollten mindestens 15-18 Wiederholungen möglich sein. Ebenfalls raten wir von Wettkampfsport ab. Für sehr sportliche Patienten und Patientinnen bieten wir auch eine interdisziplinäre Beratung mit unseren Sport-Kardiologen und -Kardiologinnen an.
- Allgemeine Verhaltensmassnahmen: es gibt prinzipiell nichts, was die betroffenen Patienten und Patientinnen unternehmen können, um die Entwicklung der Krankheit zu verhindern. Durch einen Nikotinstop und gemässigten Alkoholkonsum können die Patienten und Patientinnen aber dafür sorgen, dass ihr Herz nicht zusätzlich Schaden nimmt z.B. durch einen Herzinfarkt, der durch Rauchen begünstigt wird. Auch raten wir stark ab vom Konsum von Substanzen wie Kokain, LSD, etc. da diese Rhythmusstörungen auslösen können.
Verlauf und Prognose bei hypertropher Kardiomyopathie
Der Verlauf und die Prognose bei hypertropher Kardiomyopathie sind natürlich individuell unterschiedlich und können nicht im Detail für den einzelnen Patienten oder die einzelne Patientin vorhergesagt werden. Wie oben beschrieben, können vielfältige Komplikationen auftreten, die für den einzelnen Patienten oder die einzelne Patientin nachhaltige Auswirkungen haben kann.
Man darf aber sagen, dass die Lebenserwartung durch den Einsatz der obigen Therapiemöglichkeiten inklusive Defibrillatoren, invasiven Eingriffen bis hin zur Herztransplantation in seltenen Fällen sich in den letzten Jahrzehnten derjenigen der Normalbevölkerung angeglichen hat. Somit gehört die hypertrophe Kardiomyopathie mittlerweile zwar zu den nicht heilbaren, aber gut behandelbaren Herzmuskelerkrankungen.
Die Erstevaluation erfolgt bei uns in der Regel im Rahmen eines ambulanten Termins in unserer Spezial-Sprechstunde für hypertrophe Kardiomyopathien. Dabei ist eine ausführliche Erhebung der Krankengeschichte (Anamnese) des Patienten oder der Patientin, wie auch deren Familien zentral. Deshalb ist es vorteilhaft, wenn die Patienten und Patientinnen gut Bescheid wissen und gegebenenfalls auch Unterlagen mit zum Sprechstundentermin bringen. Neben der körperlichen Untersuchung führen wir routinemässig eine Herzstromkurve (EKG), einen Bluttest, einen Belastungstest und einen Ultraschall des Herzens (transthorakale Echokardiographie) durch. Zudem verordnen wir den Patienten und Patientinnen ein Langzeit-EKG (24-48h) und veranlassen eine Magnetresonanztomographie (MRI) des Herzens, wenn dies noch nie durchgeführt wurde.
Das sind viele Untersuchungen, die es uns aber erlauben, einerseits die Diagnose einzugrenzen – handelt es sich wirklich um eine hypertrophe Kardiomyopathie oder müssen wir an andere Ursachen der Herzmuskelerkrankung denken? – Andererseits erlauben sie es uns, eine Einschätzung über den Schweregrad der Erkrankung und insbesondere auch über das Risiko von Rhythmusstörungen durchzuführen.
Basierend auf diesen Untersuchungen können wir Empfehlungen für allfällige weiterführende Abklärungen machen, die wir beispielsweise benötigen, wenn wir eine Stoffwechselstörung vermuten (z.B. Knochenszintigraphie bei Verdacht auf Amyloidose). Zusätzlich ermöglichen uns diese Untersuchungen, optimale medikamentöse Therapien vorzuschlagen und zu beurteilen, welcher Patient oder welche Patientin ein deutlich erhöhtes Risiko für einen plötzlichen Herztod hat und daher von der Implantation eines Defibrillators profitieren würde.
Eine Erstevaluation inklusive Herz-MRI dauert bei uns mit allen Untersuchungen ungefähr einen halben Tag, ohne Herz-MRI zirka drei Stunden.
Weiterführende Abklärungen beinhalten bei uns die Möglichkeit einer genetischen Abklärung im Rahmen unserer kardiogenetischen Sprechstunde da es sich bei der hypertrophen Kardiomyopathie ja meist um eine genetische Krankheit handelt. Wichtig in diesem Zusammenhang ist, dass ein klinisches Familienscreening mit EKG und Ultraschall des Herzens bei allen erstgradigen Verwandten sehr empfohlen wird. Ziel dieser Screening-Untersuchungen ist es, die Krankheit frühzeitig erkennen zu können, um im Idealfall einen plötzlichen Herztod als Erstmanifestation der Krankheit verhindern zu können. Wenn sich in der Familie im Rahmen der genetischen Abklärung eine krankheitsverursachende Genveränderung findet, können selbstverständlich auch die Familienmitglieder auf diese Genveränderung hin untersucht werden.
Bei stabilen Patienten und Patientinnen empfehlen wir jährliche Verlaufskontrollen mit klinischer Beurteilung, EKG, Bluttest, Ultraschall des Herzens, Belastungstest und Langzeit-EKGs, um frühzeitig allfällige Komplikationen erkennen und behandeln zu können. Das Herz-MRI wiederholen wir im Abstand von drei bis fünf Jahren, da uns dies wertvolle Zusatzinformationen zusätzlich zum Ultraschall gibt.
Bei Patienten und Patientinnen, die trotz ausgebauter medikamentöser Therapie unter einer relevanten Einengung des Ausflusstrakts der linken Kammer leiden, bieten wir selbstverständlich die Evaluation und Durchführung der invasiven Therapiemöglichkeiten wie Septalalkoholablation und chirurgische Myektomie an.
Selbsthilfegruppen
Der Austausch mit Gleichbetroffenen kann bei der Bewältigung einer Krankheit eine grosse Unterstützung sein. Beratung auf der Suche nach einer geeigneten Selbsthilfegruppe erhalten Sie bei Selbsthilfe Zürich. Selbsthilfe Zürich und das Universitätsspital Zürich sind Kooperationspartner im nationalen Projekt «Gesundheitskompetenz dank selbsthilfefreundlicher Spitäler».